Aritalab:Lecture/Bioinformatics/Homology
Contents |
配列の相同性
進化の観点から似ている配列どうしを相同である (homologous) といいます。配列の相同性 (homology) という概念は、単なる類似性 (similarity) とは異なります。進化的に関係なくたまたま似ている場合は、相同であるとは言いません。配列比較をおこなうときに、生物学的に意味のある類似性を見出すことは重要なテーマです。
以下の 2 つのDNA配列 (27塩基) は相同でしょうか?
| g | c | t | a | g | g | a | t | c | a | c | g | g | c | c | a | t | g | g | c | a | a | g | c | g | c | g |
| a | a | t | t | g | a | a | g | g | a | t | t | g | c | t | c | g | g | a | t | a | a | t | c | g | c | c |
2 つの塩基がランダムだとすると、DNA配列は 1/4 の確率で一致し、3/4 の確率で不一致になります。ですから 12 塩基が一致するのはランダムな配列よりも似ているように思えます。ただし、上の揃え方では連続して一致する部分配列は長くても 3 つしか続きません。これを似ていると言ってよいのでしょうか。ここでは、相同性を判断するための様々な「指標」を考えましょう。
配列の組成、GC含量
配列がランダムであるかどうかの単純な指標は、塩基の組成をみることです。例1 の塩基配列組成は以下のようになっています。
|
|
長さが 27 塩基の場合、a, c, g, t のそれぞれは 6 ∼ 8 個ずつありそうなものですが、上の配列は t が 3 個しかありません。これはどのくらいの確率で生じる現象なのでしょうか。話を簡単にするため、gc 含量という概念を使います。塩基配列は常に相補鎖があり、a は t と、g は c と対合しています。ですから単純に t だけの量を議論するのは不正確で、a + t, g + c を比較します。
全部で 27 箇所に、at または gc がランダムに配置されるとき、平均して 13 または 14 箇所が at になると考えられます。では at が 9 箇所にしか現れない確率は、その平均的な場合に比較してどれくらい珍しいのでしょうか。それは二項係数の比を求めればわかります。
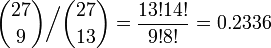
ランダムな配列と大差ないことがわかります。同様の計算で、もし at が 5 箇所にしか出てこない場合は、平均的な場合に比して 0.004 だとわかります。(稀といえるでしょう。)
最長共通部分列
配列が与えられた時、共通して現れる部分文字列(連続する必要はない)のうち、最長のものを求めるアルゴリズムを考えてみましょう。これを最長共通部分列 (LCS: longest common subsequence) と呼び、文字列 s と t を与えられたとき、それらの LCS を LCS(s, t) と書きます。
漸化式
文字列 s と t が 1 文字ずつ長くなって、それぞれ sx, ty になったとします(x, y はアルファベット 1 文字)。このとき、以下の等式が成り立ちます。(ちょっとわかりにくいですが、LCS(s,t)+x とは s と t の最長共通部分列に x を付加したものを意味しています。)
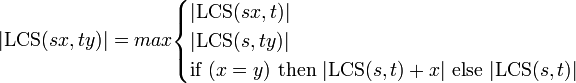
当たり前ですが s, t が長さ 0 の場合、LCS(x,y) = if (x = y) then x else "" となっています。最後の if then だけでなく、LCS(sx, t) などが必要な理由は、新しく付け加えた文字 x, y のどちらか片方だけを LCS として使うことを許すためです。
スコア行列とアルゴリズム
この式が理解できたら、動的計画法 (dynamic programming) という方法を使って LCS を計算できます[1]。例1の文字列から最初の 9 文字だけを取り出して考えましょう。途中計算を書き込む表を用意し、2 本の配列をそれぞれ第 1 行と第 1 列に配置します。この表の各セルに数字を埋めていきますが、数字は文字列の先頭からそのセルの行と列までで構成される部分文字列における LCS の長さを意味します。初期状態として、長さ 0 の文字列における LCS を意味する 0 を埋めてあります。この表をスコア行列と呼びます。
|
|
|
|
列方向の最初の文字 a と、行方向の文字列の比較を考えてみましょう。行方向の文字列で a が 1 箇所出てくれば、それ以降の文字がなんであっても LCS は 1 になります。つまり LCS(a, gcta) = LCS(a, gctaggatc) = a です。したがって、000011111 と数字を埋めていきます。 列方向に最初の 2 文字 (aa) を考えるとき、行方向の文字列で a が 2 回目に出てくる所で LCS は 2 になります。つまり LCS(aa, gctagga) = aa です。対応する LCS の長さを埋めていくと、数字は 000111222 となります。 列方向に 3 文字を (aat) をとるとき、LCS(aat, gcta) をどう埋めるべきでしょうか。LCS は a または t のどちらでもよく、長さは 1 になります。では LCS(aat, gctaggat) はどうでしょうか。LCS は aat になり 3 です。 各セルの値を埋めるときは、左隣、真上、左上の 3 箇所のセルの値だけをみれば良いことに気づきます。左隣の値、真上の値、それからセルに対応する文字が一致している場合は左上のセルの値 + 1 、そうでない場合は左上のセルの値のうち、最大値をとって埋めていけばよいのです。これを数式で表すと以下のようになります。
|
|
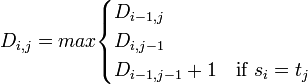
トレースバック
|
これを続けてセルを全部埋めると左のようになります。右下の数字は 4 で、実際に LCS(aattgaagg, gctaggatc) = tagg (or gagg) です。LCS文字列 tagg を求めるには、一番右下の数字 4 が生成された経緯をたどります。この処理をトレースバック (trace back) といいます。 数字の 4 は、最下段で太字で示したセルで初めて現れました。これは文字 g の一致 (aattgaagg, gctaggatc) に対応します。その左上のセルは 3 で、このセルも同じく g の一致により更に左上のセルから生成されています。4 から左上にたどると agg という文字列の一致 (aattgaagg, gctaggatc) に対応します。太字の数字 2 の左上には 1 がありますが、この値はこのセルで生じたものではありません。左隣か上のセルに由来しています。1 が初めて生じたセルを探すと太字で示した 1 にたどり着きます。トレースバック道筋は一通りとは限らず、このどちらをとっても正解です。この例題では、トレースバックによって tagg または gagg という LCS を求められます。 |
|
アライメントスコア
LCS アルゴリズムは一致した文字数のみを扱っていて、一致しない文字は全く考慮しません。しかし上の例で考えた gagg という LCS でも、
|
|
|
といった文字列の並べ方があります。記号 - はギャップと呼び、片側の塩基が抜けていること(または塩基が挿入されていること)を意味します。塩基配列の進化という観点からは、上の 3 つはいずれも異なる意味を持ちます。左の 2 例はギャップが 1 箇所しかありませんが、右の例はギャップが両方の配列に入っています。また左の 2 例は塩基のミスマッチを含みますが、右の例は含みません。
生物由来の配列比較、つまりアライメント (alignment) をおこなう際は、マッチ、ミスマッチ、ギャップのそれぞれにスコアを割り振り、アライメントスコアとして
(マッチ数) − μ(ミスマッチ数) − σ(ギャップスペース数)
を考えます。ここで μ, σ は 1 塩基あたりのペナルティスコアで、マッチのスコアを 1 とした相対値で表します。LCSアルゴリズムにおける動的計画法のスコア s は以下のように記せます。
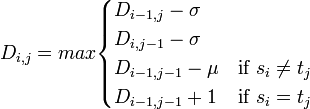
大域・局所アライメント
与えられた 2 つの配列全体を一致させる並べ方を大域 (global) アライメントと呼びます。同じファミリーに属するタンパク質配列の比較や、進化的に近いことが知られている配列同士の詳細な比較に用います。これに対して、与えられた配列をデータベース中で検索する場合を考えてみましょう。データベース中のごく一部分とどの程度似るかが問題になります。これを局所 (local) アライメントと呼び、大域アライメントとは以下の関係にあります。
局所アライメント = 与えられた2つの配列それぞれの全ての部分配列のペア中で、大域アライメントのスコアを最大にするもの
すべての部分配列のペアを考える部分が面倒に思えますが、実際は動的計画法において初期設定を変更するだけで大域アライメントと同様に計算できます。 具体的なアルゴリズムを別ページでみていきましょう。
- 参考
- ↑ 動的計画法 (dynamic programming) とはプログラミングと全く関係が無いのですが、アルゴリズムが命名されたのが1940年代(コンピュータ言語が生まれる前)なのでそのまま呼ばれています。