Aritalab:Lecture/Bioinformatics/Phylogeny
m (→近隣結合法 NJ) |
(→進化系統樹) |
||
| (8 intermediate revisions by one user not shown) | |||
| Line 2: | Line 2: | ||
配列進化を考える際は、配列にランダムに変化が入る[[Aritalab:Lecture/NetworkBiology/Markov_Chains|マルコフモデル]]を用います。配列の置換やギャップは[[Aritalab:Lecture/NetworkBiology/Markov_Chains/Birth-death_Process|出生死亡過程]]としてモデルします<ref>JL Thorne, H Kishino, J Felsenstein (1992) "Inching toward reality: an improved likelihood model of sequence evolution" J Mol Biol 34, 3-16</ref>。 | 配列進化を考える際は、配列にランダムに変化が入る[[Aritalab:Lecture/NetworkBiology/Markov_Chains|マルコフモデル]]を用います。配列の置換やギャップは[[Aritalab:Lecture/NetworkBiology/Markov_Chains/Birth-death_Process|出生死亡過程]]としてモデルします<ref>JL Thorne, H Kishino, J Felsenstein (1992) "Inching toward reality: an improved likelihood model of sequence evolution" J Mol Biol 34, 3-16</ref>。 | ||
| − | + | <!---- | |
===置換速度行列=== | ===置換速度行列=== | ||
| − | + | 塩基 A, C, G, T がそれぞれ他の塩基に変化する速度を 4 x 4 行列の形式で表したものを置換速度行列と呼びます。 | |
一番単純なのは Junks-Cantor (JC) モデルで、全ての塩基が等確率で別の塩基に変化すると仮定します。 | 一番単純なのは Junks-Cantor (JC) モデルで、全ての塩基が等確率で別の塩基に変化すると仮定します。 | ||
<math> R = \begin{bmatrix} | <math> R = \begin{bmatrix} | ||
| − | - 3 \alpha & \alpha & \alpha & \alpha \\ | + | - 3\alpha & \alpha & \alpha & \alpha \\ |
| − | \alpha & - \alpha & \alpha & \alpha \\ | + | \alpha & - 3\alpha & \alpha & \alpha \\ |
| − | \alpha & \alpha & - \alpha & \alpha \\ | + | \alpha & \alpha & - 3\alpha & \alpha \\ |
| − | \alpha & \alpha & \alpha & - \alpha | + | \alpha & \alpha & \alpha & - 3\alpha |
\end{bmatrix}</math> | \end{bmatrix}</math> | ||
===トランジションとトランスバージョン=== | ===トランジションとトランスバージョン=== | ||
| − | プリン間 ( A ⇔ G )、ピリミジン間 ( C ⇔ T ) の置換を転位 (transition) とよび、それ以外の(つまりプリンとピリミジン間の)置換を転換 (transversion) と呼びます。転位のほうが起きやすい置換のため、これを反映させたのが木村資生による 2 | + | プリン間 ( A ⇔ G )、ピリミジン間 ( C ⇔ T ) の置換を転位 (transition) とよび、それ以外の(つまりプリンとピリミジン間の)置換を転換 (transversion) と呼びます。転位のほうが起きやすい置換のため、これを反映させたのが木村資生による 2 変数モデルです。転位の置換速度を α、転換の速度を β で表します。 |
<math> R = \begin{bmatrix} | <math> R = \begin{bmatrix} | ||
| Line 26: | Line 26: | ||
塩基の置換はアミノ酸の変更につながる場合があります。同じアミノ酸をコードするコドン間の置換を同義置換 (synonymous substitution)、そうでないものを非同義置換と呼びます。 | 塩基の置換はアミノ酸の変更につながる場合があります。同じアミノ酸をコードするコドン間の置換を同義置換 (synonymous substitution)、そうでないものを非同義置換と呼びます。 | ||
| + | ----> | ||
==進化系統樹== | ==進化系統樹== | ||
| Line 31: | Line 32: | ||
生物の進化にそった種分岐を樹状に表現したものが系統樹 (phylogenetic tree) です。樹の葉(末端部分)が現存する生物種にあたり、遡った頂点ほど祖先にあたる(現存しない)生物種を意味します。根つき (rooted) で描く方法と、根を仮定しない (unrooted) 方法があり、根を求めるには、対象とする生物種とおおきくかけ離れた種をひとつ加えます。これを外群 (outgroup) と呼び、この種と残りすべてを結ぶ頂点を根とみなします。 | 生物の進化にそった種分岐を樹状に表現したものが系統樹 (phylogenetic tree) です。樹の葉(末端部分)が現存する生物種にあたり、遡った頂点ほど祖先にあたる(現存しない)生物種を意味します。根つき (rooted) で描く方法と、根を仮定しない (unrooted) 方法があり、根を求めるには、対象とする生物種とおおきくかけ離れた種をひとつ加えます。これを外群 (outgroup) と呼び、この種と残りすべてを結ぶ頂点を根とみなします。 | ||
| − | + | 系統樹を作成するには、アライメント等で求めた配列間の距離から作る場合と、配列そのものを用いて作る場合の二通りがあります。距離から作る場合の代表例が NJ法、配列から作る場合の代表例が MP法です。 | |
===距離から作る場合=== | ===距離から作る場合=== | ||
| Line 39: | Line 40: | ||
# 距離が最小のペア i, j を選ぶ (距離を d<sub>i,j</sub> とします) | # 距離が最小のペア i, j を選ぶ (距離を d<sub>i,j</sub> とします) | ||
# 生物種 i と j を、距離 d<sub>i,j</sub>/ 2 に位置する分岐点 u を用いてつなぎ、この点を新しい種とみなす | # 生物種 i と j を、距離 d<sub>i,j</sub>/ 2 に位置する分岐点 u を用いてつなぎ、この点を新しい種とみなす | ||
| − | # 他の種 k | + | # 他の種 k から u までの距離を d<sub>k,u</sub> = (d<sub>k,i</sub> + d<sub>k,j</sub>)/2 (つまり i までと j までの距離の平均)として再計算 |
| − | # | + | # このステップを、全生物種がまとまるまで繰り返す |
| + | |||
| + | UPGMAは配列進化の速度が一定であると仮定しているため、ステップ 1 では i , j から等距離のところに分岐点 u を作成します。これを繰り返すと、各分岐点から葉頂点(各生物種)までの長さが揃った有根系統樹が作成されます(一番最後に作成した分岐点が最古の生物種)。 | ||
| − | + | 進化速度一定という仮定は正しいとは思えません。その理由はUPGMA法がタンパク質などの電気泳動パターンを分類するために開発された手法だからです。計算は早くて便利ですが、UPGMA法を進化系統樹の作成法として広めたのが良くなかったと思います。教科書には載っていますが実際には使われない(使ってはいけない)手法です。 | |
====近隣結合法 NJ==== | ====近隣結合法 NJ==== | ||
| − | NJ は Neighbor Joining | + | NJ は Neighbor Joining 法の略称で、日本の斎藤成也と根井正利によって無根の進化系統樹を作成するために1987年に発表されました。ソフトウェアは、Felsenstein グループが1980年から [http://evolution.genetics.washington.edu/phylip.html Phylip]を公開し、よく利用されています。 |
# 各生物種 i に対し、他からの距離の総和 <math>D_i = \textstyle \sum^n_{k=1} d_{i,k}</math> を求める | # 各生物種 i に対し、他からの距離の総和 <math>D_i = \textstyle \sum^n_{k=1} d_{i,k}</math> を求める | ||
# <math>\textstyle d_{i,j} - \frac{(D_i + D_j)}{n-2}</math> を最小にするペア i , j を選ぶ | # <math>\textstyle d_{i,j} - \frac{(D_i + D_j)}{n-2}</math> を最小にするペア i , j を選ぶ | ||
| − | # 生物種 i と j を<br/> i から <math>\textstyle \frac{d_{i,j | + | # 生物種 i と j を<br/> i から <math>\textstyle \frac{1}{2}(d_{i,j} - \frac{(D_i - D_j)}{(n-2)})</math><br/> j から <math>\textstyle \frac{1}{2} (d_{i,j} - \frac{(D_j - D_i)}{(n-2)})</math> の分岐点 u を用いてつなぐ |
| − | # 他の種 k からの距離を | + | # 他の種 k からの距離を <math>\textstyle d_{k,u} = (d_{k,i} + d_{k,j} - d_{i,j})/2 </math> として再計算 |
# このステップを全生物種がまとまるまで繰り返す | # このステップを全生物種がまとまるまで繰り返す | ||
| − | ステップ 2 | + | ステップ 2 の部分では、生物種全てで星状の系統樹を作ったときに一番中心に近い生物種を選んでいます。ステップ 3 の部分では、i と j をまとめる点として新しい頂点 u を設定しますが、その i と j からの距離は d<sub>u,i</sub> + d<sub>u,j</sub> = d<sub>i,j</sub> を満たし、D<sub>i</sub> と D<sub>j</sub> の差を考慮して設定されています。 |
| + | |||
| + | 距離が近いものから順次つなぐために全枝長を最小にする系統樹が得られる保証はありませんが、実用上はこれで十分です。 | ||
===配列を直接作る場合=== | ===配列を直接作る場合=== | ||
| Line 62: | Line 67: | ||
==ツール、ソフトウェア== | ==ツール、ソフトウェア== | ||
| − | + | ナショナルバイオデータベースセンターによるツールの紹介は [http://stga.biosciencedbc.jp/cgi-bin/link.cgi?category=4100 こちら]。 | |
| + | 国内で開発されるソフトとして、実験医学増刊[http://www.yodosha.co.jp/jikkenigaku/book/9784758103176/ 「使えるデータベース・ウェブツール」]では、MEGA を薦めています。 | ||
;参考 | ;参考 | ||
<references/> | <references/> | ||
Latest revision as of 11:15, 20 August 2012
Contents |
[edit] 配列の進化
配列進化を考える際は、配列にランダムに変化が入るマルコフモデルを用います。配列の置換やギャップは出生死亡過程としてモデルします[1]。
[edit] 進化系統樹
生物の進化にそった種分岐を樹状に表現したものが系統樹 (phylogenetic tree) です。樹の葉(末端部分)が現存する生物種にあたり、遡った頂点ほど祖先にあたる(現存しない)生物種を意味します。根つき (rooted) で描く方法と、根を仮定しない (unrooted) 方法があり、根を求めるには、対象とする生物種とおおきくかけ離れた種をひとつ加えます。これを外群 (outgroup) と呼び、この種と残りすべてを結ぶ頂点を根とみなします。
系統樹を作成するには、アライメント等で求めた配列間の距離から作る場合と、配列そのものを用いて作る場合の二通りがあります。距離から作る場合の代表例が NJ法、配列から作る場合の代表例が MP法です。
[edit] 距離から作る場合
[edit] 平均距離法 UPGMA
UPGMA は Unweighted Pair Group Method with Arithmetic Means の略です。複数生物種間の進化距離を与えられた時、以下の手順で系統樹を作成します。
- 距離が最小のペア i, j を選ぶ (距離を di,j とします)
- 生物種 i と j を、距離 di,j/ 2 に位置する分岐点 u を用いてつなぎ、この点を新しい種とみなす
- 他の種 k から u までの距離を dk,u = (dk,i + dk,j)/2 (つまり i までと j までの距離の平均)として再計算
- このステップを、全生物種がまとまるまで繰り返す
UPGMAは配列進化の速度が一定であると仮定しているため、ステップ 1 では i , j から等距離のところに分岐点 u を作成します。これを繰り返すと、各分岐点から葉頂点(各生物種)までの長さが揃った有根系統樹が作成されます(一番最後に作成した分岐点が最古の生物種)。
進化速度一定という仮定は正しいとは思えません。その理由はUPGMA法がタンパク質などの電気泳動パターンを分類するために開発された手法だからです。計算は早くて便利ですが、UPGMA法を進化系統樹の作成法として広めたのが良くなかったと思います。教科書には載っていますが実際には使われない(使ってはいけない)手法です。
[edit] 近隣結合法 NJ
NJ は Neighbor Joining 法の略称で、日本の斎藤成也と根井正利によって無根の進化系統樹を作成するために1987年に発表されました。ソフトウェアは、Felsenstein グループが1980年から Phylipを公開し、よく利用されています。
- 各生物種 i に対し、他からの距離の総和
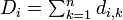 を求める
を求める
-
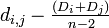 を最小にするペア i , j を選ぶ
を最小にするペア i , j を選ぶ
- 生物種 i と j を
i から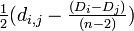
j から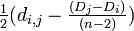 の分岐点 u を用いてつなぐ
の分岐点 u を用いてつなぐ
- 他の種 k からの距離を
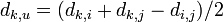 として再計算
として再計算
- このステップを全生物種がまとまるまで繰り返す
ステップ 2 の部分では、生物種全てで星状の系統樹を作ったときに一番中心に近い生物種を選んでいます。ステップ 3 の部分では、i と j をまとめる点として新しい頂点 u を設定しますが、その i と j からの距離は du,i + du,j = di,j を満たし、Di と Dj の差を考慮して設定されています。
距離が近いものから順次つなぐために全枝長を最小にする系統樹が得られる保証はありませんが、実用上はこれで十分です。
[edit] 配列を直接作る場合
[edit] 最大節約法 MP
MP 法は maximum parsimony を意味し、最節約法とも呼ばれます。塩基の置換数を最低にするように、最尤法 (maximum likelihood method) で系統樹を作成します。計算時間が長いため、少数の配列についてしか適用できません。
[edit] ツール、ソフトウェア
ナショナルバイオデータベースセンターによるツールの紹介は こちら。 国内で開発されるソフトとして、実験医学増刊「使えるデータベース・ウェブツール」では、MEGA を薦めています。
- 参考
- ↑ JL Thorne, H Kishino, J Felsenstein (1992) "Inching toward reality: an improved likelihood model of sequence evolution" J Mol Biol 34, 3-16