Aritalab:Lecture/Bioinformatics/MotifFinding
(→モチーフ発見問題) |
m |
||
| (7 intermediate revisions by one user not shown) | |||
| Line 5: | Line 5: | ||
===プロファイル行列とコンセンサス=== | ===プロファイル行列とコンセンサス=== | ||
| − | 固定長 ( | + | 固定長 (k とします) の配列を n 本そろえて n × k 行列の形に表現し、各列における各塩基配列の出現頻度を記した 4 × k 行列をプロファイルと呼びます。 |
| − | プロファイルの ( | + | プロファイルの ( i, j ) 要素 ( i ∈ {A,C,G,T}, j ∈ Z) には、もとの行列で塩基 i が出現する確率が入ります。 |
それぞれの位置で代表的な塩基が定まる場合、それをコンセンサス配列と呼びます。 | それぞれの位置で代表的な塩基が定まる場合、それをコンセンサス配列と呼びます。 | ||
| Line 79: | Line 79: | ||
==モチーフ発見問題== | ==モチーフ発見問題== | ||
| − | ここでは長いDNA配列 | + | ここでは長いDNA配列 n 本と正の整数 k を与えられたとき、配列それぞれの位置 '''s''' = (s<sub>1</sub>, s<sub>2</sub>, ... s<sub>n</sub>) にから長さ k の部分配列を取り出してできるプロファイル行列を考えます。 |
| − | : '''問題.''' スコア関数 S('''s''', DNA) = − <Big>Σ<sup> | + | : '''問題.''' スコア関数 S( '''s''' , DNA) = − <Big>Σ<sup>k</sup><sub>j</sub> Σ<sub>i</sub><sup>A,T,G,C</sup></big> p<sub>i,j</sub> log p<sub>i,j</sub> を最小化する位置の配列 '''s''' を求めよ。 |
これをモチーフ発見問題といいます。スコア関数は − <big>ΣΣ</big> p log p の形をとっており、p<sub>ij</sub> はポジション j における塩基 i の確率を表しています。− p log p の総和をプロファイルのエントロピーといいます。 | これをモチーフ発見問題といいます。スコア関数は − <big>ΣΣ</big> p log p の形をとっており、p<sub>ij</sub> はポジション j における塩基 i の確率を表しています。− p log p の総和をプロファイルのエントロピーといいます。 | ||
| − | このエントロピーとは[[Aritalab:Lecture/Basic/Entropy|シャノンの平均情報量]] | + | このエントロピーとは[[Aritalab:Lecture/Basic/Entropy|シャノンの平均情報量]]を意味し、分布の偏りを示す指標です。全ての塩基が等確率で出てくるときに平均情報量は最も大きく、塩基の出現確率に偏りがあればあるほど、平均情報量は小さくなります。 |
| − | === | + | * 完全にランダムな場合 (情報量は大きい) |
| − | 長さ L の DNA 配列が n 本与えられたとき、長さ | + | : − Σ<sup>k</sup>Σ<sup>ACGT</sup> p log p = − 4 k * { (1/4) * (−2)} = 2 k |
| + | : k ビットの情報源が持ちうるエントロピーの最大値は k です。DNAは4文字あるために各位置が 2 ビットの情報を持ち得て 2k になります。もし4文字のうち2文字だけが50%ずつ利用される場合、情報量は k になります。 | ||
| + | |||
| + | * 完全に一致する場合(情報量は小さい) | ||
| + | : − Σ<sup>k</sup>Σ<sup>ACGT</sup> p log p = − k * { log 1 } = 0 | ||
| + | : 完全一致の場合、情報量は 0 になります。 | ||
| + | |||
| + | このスコア関数をハミング距離にしても本質的な問題は変わりません。完全にランダムな場合はハミング距離の総和が最大になり、完全一致の場合はハミング距離の総和が最小になります。 | ||
| + | |||
| + | ===モチーフ候補探しの効率=== | ||
| + | |||
| + | 長さ L の DNA 配列が n 本与えられたとき、長さ k のモチーフを選ぶ組み合わせの数は (L - k + 1)<sup>n</sup> になります。それぞれのポジションについてプロファイル行列を kn 時間で作成していると、全体で | ||
| + | |||
| + | (L - k + 1)<sup>n</sup> × k n = O(k n L<sup>n</sup>) 時間 | ||
| + | |||
| + | かかります。これは大変無駄の多いアプローチです。代案として、コンセンサス配列を全て求めるアプローチが考えられます。例えば、プロファイル行列において一番頻度の高い文字を採用したコンセンサスでエントロピーが k 以上のものを求める問題を考えます。コンセンサス配列の数は 4<sup>k</sup> 個あり、それぞれに対して nL 配列の中に類似配列が存在するか否かを判断するのに knL 時間かかります。 | ||
| + | |||
| + | 4<sup>k</sup> × k n L = O(k n L 4<sup>k</sup>) 時間 | ||
| + | |||
| + | こちらも無駄の多いアプローチに見えますが、n が大きかったり k が小さいときには有効なことがわかります。 | ||
| + | |||
| + | ===枝刈り=== | ||
| + | |||
| + | L<sup>n</sup> や 4<sup>k</sup> という組み合わせの数は膨大で、全てのパターンをしらみつぶしに計算していると無駄も多そうです。あらかじめ設定した閾値などを用いて無駄な手続きを省略する手法を枝刈り (branch-and-bound) と呼びます。こうした枝刈りでは、残念ながら最悪計算量を下げることはできません。 | ||
| + | |||
| + | ; 例. プロファイル行列作りにおける枝刈り | ||
| + | |||
| + | 長さ k のモチーフと呼べるプロファイルの各ポジションにおけるエントロピーを、k 以下としてみましょう。 各位置のエントロピーが 1 以下であるためには少なくとも半分以上の文字が同一でなくてはなりません。従って、n 本の配列に対して (L - k + 1)<sup>n</sup> 通りの組み合わせを試すときに、途中で残りの文字がどのようになってもエントロピー 1 を達成できないことがわかれば、その時点で次の候補へ移ることができます。 | ||
| + | |||
| + | 例えば i 本目の比較で目標のエントロピー基準を達成できないことがわかれば、i + 1 本目以降の (L - k + 1)<sup>n - i</sup> 通りをスキップできます。 | ||
| + | |||
| + | ==ギブスサンプリング== | ||
| + | |||
| + | ギブスサンプリングはもともと統計力学などで用いられてきた手法ですが、1993年に Charles Lawrence らが Science 誌に発表した論文<ref>CE Lawrence, SF Altschul, MS Boguski, JS Liu, AF Neuwald, JC Wootton "Subtle Sequence Signals: A Gibbs Sampling Strategy for Multiple Alignment" Science 262(5131), 208-214, 1993</ref>がきっかけで分子生物学分野でも急速に用いられるようになりました。この論文ではヘリックス・ターン・ヘリックス (HTH) モチーフなどを効率よく発見する手法が紹介されています。 | ||
| + | |||
| + | ===アルゴリズム=== | ||
| + | |||
| + | n 本の配列それぞれに何らかの重要なモチーフがあるものの、場所がわからない場合を想定します。 | ||
| + | ギブスサンプリングは以下の手続きによってモチーフ(またはシグナル)位置をみつける確率的手法です。モチーフの長さは予め与えておく必要があります。(ここでは長さ k とします。) | ||
| + | |||
| + | # それぞれの配列からモチーフ位置 s = ( s<sub>1</sub>, s<sub>2</sub>, s<sub>3</sub>, ... s<sub>n</sub> ) をランダムに選ぶ | ||
| + | # 配列 1 本をランダムに抜きとり(i 番目とします)、残りの n − 1 配列から s に従って長さ k のプロファイル P を作成する | ||
| + | # 抜いた i 番目の配列において、配列の各位置におけるプロファイル P のスコア ( p<sub>1</sub>, p<sub>2</sub>, ... , p<sub>n-k+1</sub> ) を計算する | ||
| + | # スコアを確率分布とみなし、その中から配列 i に対するモチーフ位置 s<sub>i</sub> を決めて s を更新する | ||
| + | # 更新によって値が変化しなくなるまで(収束するまで)繰り返す | ||
| + | |||
| + | もし s がばらばらな配列要素を指していたらプロファイルは平均的なものになるため、更新されるモチーフ位置はほとんどランダムに決定されます。しかし何らかの正しいモチーフに近いプロファイルが形成された途端、プロファイルは正確にモチーフを選び出すようになって収束します。最適解を返す保証はありませんが、実用上は大変便利なことが知られています。 | ||
| + | |||
| + | ===スコア関数=== | ||
| + | |||
| + | アミノ酸配列のモチーフを求める実用プログラムでは、プロファイル P のスコア関数 F を以下のように設定します。 | ||
| + | <center> | ||
| + | <math>F = \sum^{k}_{i=1}\sum^{20}_{j=1}\frac{c_{i,j}}{n-1} \log \frac{c_{i,j} + b_j}{\bar{c}_{i,j} + b_j}</math> | ||
| + | </center> | ||
| + | ここで j は 20 種のアミノ酸に対応する番号で、i は長さ k のモチーフにおける位置を示しています。記号 <math>c_{i,j}, \bar{c}_{j}</math> はそれぞれ、モチーフ位置 i におけるアミノ酸 j の出現回数と、モチーフ以外の部分全体にでてくるアミノ酸 j の回数(バックグラウンド回数)です。また <math> b_j </math> は擬似カウント (pseudocount) と呼ぶ値で、実際のアミノ酸頻度による値を "なます" 効果を持つパラメータです。 | ||
| + | |||
| + | 基本は <math>\sum - p \log p </math> という平均情報量を計算します。しかし各アミノ酸は均等な割合で出てくるわけではないので単純な頻度である <math>\textstyle p = \frac{c_{i,j}}{(n-1)}</math> の代わりに背景となるアミノ酸頻度との比 <math>\textstyle \frac{c_{i,j}}{\bar{c}_{j}}</math> をアミノ酸 j の情報量とし、さらに擬似カウントをつけ足しています。 | ||
| + | |||
| + | |||
| + | ===実用化へのテクニック=== | ||
| + | ;モチーフの左右へのズレ | ||
| + | 3本の配列で正解のモチーフがそれぞれ位置 a, b, c から始まる場合、たまたま a+2, b+2 の位置をアルゴリズムが見つけたら c+2 をモチーフの開始位置にしてしまいます。これを避けるには、モチーフのプロファイルを作成する際に位置を一定間隔だけ左右にずらし、よりよいモチーフが得られないかどうかを判定します。 | ||
| + | |||
| + | ;長さ k を自動的に決定する工夫 | ||
| + | 通常はモチーフの長さ k をあらかじめ知ることができません。しかし、k を自由に設定できるようにすると、その値が長いほどプロファイルのスコアが高くなって不必要に長いモチーフを探してしまいます。これを防ぐには、プロファイルのスコアから k に比例する値を引いて k の長さを調節させます。ここは引く値を経験的に求める必要があり、スコア関数 F との兼ね合いもあるので理論的に指針を出すことができません。 | ||
| + | |||
| + | ;参考 | ||
| + | <references/> | ||
Latest revision as of 20:40, 18 December 2011
Contents |
[edit] モチーフ
DNA配列の中で転写制御部位や生体分子の結合部位など、決まったパターンを持つ部分配列をモチーフと呼びます。 決まったパターンといっても全く同じ部分配列が存在することは稀で、多くは「ほとんど」一致する部分配列となります。 モチーフを表現するのに、正則表現やプロファイル行列がよく用いられます。
[edit] プロファイル行列とコンセンサス
固定長 (k とします) の配列を n 本そろえて n × k 行列の形に表現し、各列における各塩基配列の出現頻度を記した 4 × k 行列をプロファイルと呼びます。 プロファイルの ( i, j ) 要素 ( i ∈ {A,C,G,T}, j ∈ Z) には、もとの行列で塩基 i が出現する確率が入ります。
それぞれの位置で代表的な塩基が定まる場合、それをコンセンサス配列と呼びます。
- 例
|
⇒ |
|
⇒ |
|
[edit] モチーフ発見問題
ここでは長いDNA配列 n 本と正の整数 k を与えられたとき、配列それぞれの位置 s = (s1, s2, ... sn) にから長さ k の部分配列を取り出してできるプロファイル行列を考えます。
- 問題. スコア関数 S( s , DNA) = − Σkj ΣiA,T,G,C pi,j log pi,j を最小化する位置の配列 s を求めよ。
これをモチーフ発見問題といいます。スコア関数は − ΣΣ p log p の形をとっており、pij はポジション j における塩基 i の確率を表しています。− p log p の総和をプロファイルのエントロピーといいます。 このエントロピーとはシャノンの平均情報量を意味し、分布の偏りを示す指標です。全ての塩基が等確率で出てくるときに平均情報量は最も大きく、塩基の出現確率に偏りがあればあるほど、平均情報量は小さくなります。
- 完全にランダムな場合 (情報量は大きい)
- − ΣkΣACGT p log p = − 4 k * { (1/4) * (−2)} = 2 k
- k ビットの情報源が持ちうるエントロピーの最大値は k です。DNAは4文字あるために各位置が 2 ビットの情報を持ち得て 2k になります。もし4文字のうち2文字だけが50%ずつ利用される場合、情報量は k になります。
- 完全に一致する場合(情報量は小さい)
- − ΣkΣACGT p log p = − k * { log 1 } = 0
- 完全一致の場合、情報量は 0 になります。
このスコア関数をハミング距離にしても本質的な問題は変わりません。完全にランダムな場合はハミング距離の総和が最大になり、完全一致の場合はハミング距離の総和が最小になります。
[edit] モチーフ候補探しの効率
長さ L の DNA 配列が n 本与えられたとき、長さ k のモチーフを選ぶ組み合わせの数は (L - k + 1)n になります。それぞれのポジションについてプロファイル行列を kn 時間で作成していると、全体で
(L - k + 1)n × k n = O(k n Ln) 時間
かかります。これは大変無駄の多いアプローチです。代案として、コンセンサス配列を全て求めるアプローチが考えられます。例えば、プロファイル行列において一番頻度の高い文字を採用したコンセンサスでエントロピーが k 以上のものを求める問題を考えます。コンセンサス配列の数は 4k 個あり、それぞれに対して nL 配列の中に類似配列が存在するか否かを判断するのに knL 時間かかります。
4k × k n L = O(k n L 4k) 時間
こちらも無駄の多いアプローチに見えますが、n が大きかったり k が小さいときには有効なことがわかります。
[edit] 枝刈り
Ln や 4k という組み合わせの数は膨大で、全てのパターンをしらみつぶしに計算していると無駄も多そうです。あらかじめ設定した閾値などを用いて無駄な手続きを省略する手法を枝刈り (branch-and-bound) と呼びます。こうした枝刈りでは、残念ながら最悪計算量を下げることはできません。
- 例. プロファイル行列作りにおける枝刈り
長さ k のモチーフと呼べるプロファイルの各ポジションにおけるエントロピーを、k 以下としてみましょう。 各位置のエントロピーが 1 以下であるためには少なくとも半分以上の文字が同一でなくてはなりません。従って、n 本の配列に対して (L - k + 1)n 通りの組み合わせを試すときに、途中で残りの文字がどのようになってもエントロピー 1 を達成できないことがわかれば、その時点で次の候補へ移ることができます。
例えば i 本目の比較で目標のエントロピー基準を達成できないことがわかれば、i + 1 本目以降の (L - k + 1)n - i 通りをスキップできます。
[edit] ギブスサンプリング
ギブスサンプリングはもともと統計力学などで用いられてきた手法ですが、1993年に Charles Lawrence らが Science 誌に発表した論文[1]がきっかけで分子生物学分野でも急速に用いられるようになりました。この論文ではヘリックス・ターン・ヘリックス (HTH) モチーフなどを効率よく発見する手法が紹介されています。
[edit] アルゴリズム
n 本の配列それぞれに何らかの重要なモチーフがあるものの、場所がわからない場合を想定します。 ギブスサンプリングは以下の手続きによってモチーフ(またはシグナル)位置をみつける確率的手法です。モチーフの長さは予め与えておく必要があります。(ここでは長さ k とします。)
- それぞれの配列からモチーフ位置 s = ( s1, s2, s3, ... sn ) をランダムに選ぶ
- 配列 1 本をランダムに抜きとり(i 番目とします)、残りの n − 1 配列から s に従って長さ k のプロファイル P を作成する
- 抜いた i 番目の配列において、配列の各位置におけるプロファイル P のスコア ( p1, p2, ... , pn-k+1 ) を計算する
- スコアを確率分布とみなし、その中から配列 i に対するモチーフ位置 si を決めて s を更新する
- 更新によって値が変化しなくなるまで(収束するまで)繰り返す
もし s がばらばらな配列要素を指していたらプロファイルは平均的なものになるため、更新されるモチーフ位置はほとんどランダムに決定されます。しかし何らかの正しいモチーフに近いプロファイルが形成された途端、プロファイルは正確にモチーフを選び出すようになって収束します。最適解を返す保証はありませんが、実用上は大変便利なことが知られています。
[edit] スコア関数
アミノ酸配列のモチーフを求める実用プログラムでは、プロファイル P のスコア関数 F を以下のように設定します。
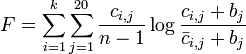
ここで j は 20 種のアミノ酸に対応する番号で、i は長さ k のモチーフにおける位置を示しています。記号 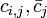 はそれぞれ、モチーフ位置 i におけるアミノ酸 j の出現回数と、モチーフ以外の部分全体にでてくるアミノ酸 j の回数(バックグラウンド回数)です。また
はそれぞれ、モチーフ位置 i におけるアミノ酸 j の出現回数と、モチーフ以外の部分全体にでてくるアミノ酸 j の回数(バックグラウンド回数)です。また 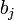 は擬似カウント (pseudocount) と呼ぶ値で、実際のアミノ酸頻度による値を "なます" 効果を持つパラメータです。
は擬似カウント (pseudocount) と呼ぶ値で、実際のアミノ酸頻度による値を "なます" 効果を持つパラメータです。
基本は 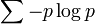 という平均情報量を計算します。しかし各アミノ酸は均等な割合で出てくるわけではないので単純な頻度である
という平均情報量を計算します。しかし各アミノ酸は均等な割合で出てくるわけではないので単純な頻度である 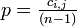 の代わりに背景となるアミノ酸頻度との比
の代わりに背景となるアミノ酸頻度との比 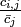 をアミノ酸 j の情報量とし、さらに擬似カウントをつけ足しています。
をアミノ酸 j の情報量とし、さらに擬似カウントをつけ足しています。
[edit] 実用化へのテクニック
- モチーフの左右へのズレ
3本の配列で正解のモチーフがそれぞれ位置 a, b, c から始まる場合、たまたま a+2, b+2 の位置をアルゴリズムが見つけたら c+2 をモチーフの開始位置にしてしまいます。これを避けるには、モチーフのプロファイルを作成する際に位置を一定間隔だけ左右にずらし、よりよいモチーフが得られないかどうかを判定します。
- 長さ k を自動的に決定する工夫
通常はモチーフの長さ k をあらかじめ知ることができません。しかし、k を自由に設定できるようにすると、その値が長いほどプロファイルのスコアが高くなって不必要に長いモチーフを探してしまいます。これを防ぐには、プロファイルのスコアから k に比例する値を引いて k の長さを調節させます。ここは引く値を経験的に求める必要があり、スコア関数 F との兼ね合いもあるので理論的に指針を出すことができません。
- 参考
- ↑ CE Lawrence, SF Altschul, MS Boguski, JS Liu, AF Neuwald, JC Wootton "Subtle Sequence Signals: A Gibbs Sampling Strategy for Multiple Alignment" Science 262(5131), 208-214, 1993